


 Le tératome sacrococcygien (TSC) est la tumeur la plus fréquente du fœtus et du nouveau-né, à la frontière entre malformation congénitale et oncologie pédiatrique, mais reste très rare avec une prévalence estimée entre 1/40.000 et 1/10.000 naissances. Il touche plus fréquemment les filles que les garçons (4:1).
Le tératome sacrococcygien (TSC) est la tumeur la plus fréquente du fœtus et du nouveau-né, à la frontière entre malformation congénitale et oncologie pédiatrique, mais reste très rare avec une prévalence estimée entre 1/40.000 et 1/10.000 naissances. Il touche plus fréquemment les filles que les garçons (4:1).
Le TSC est défini comme une tumeur appendue au coccyx et contenant des tissus provenant d’au moins deux des trois feuillets embryonnaires (endoderme, mésoderme, neurectoderme), qui peuvent être retrouvés à des degrés de maturation plus ou moins avancés. Sa genèse exacte reste encore inconnue.
Les TSC sont classés selon la classification d’Altman (I à IV) [1], en fonction de la part endo- et exopelvienne de la tumeur. Cette tumeur malformative est habituellement isolée, et bénigne lorsqu’elle est diagnostiquée en période prénatale ou néonatale.
Figure 1 : Classification de Altman des tératomes sacro-coccygiens.
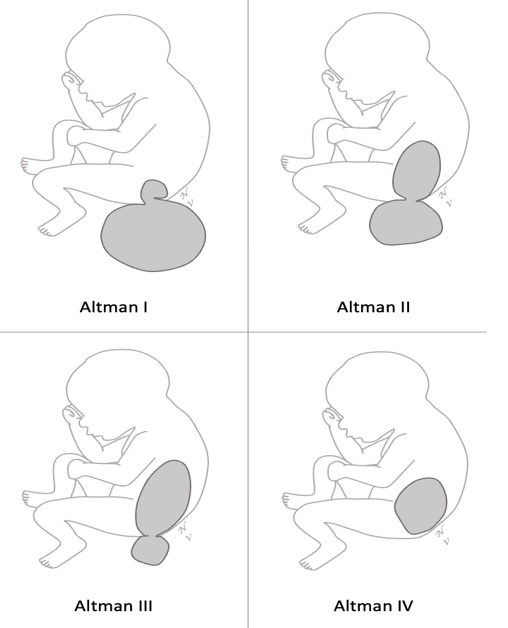
Type I : TSC majoritairement exo-pelvien avec une petite composante endopelvienne. Type II : TSC à composante endo- et exopelvienne équilibrée. Type III : TSC à prédominance endopelvienne avec petite composante exopelvienne. Type IV : TSC strictement endopelvien.
Dans sa forme syndromique, le TSC s’inscrit le plus souvent dans le syndrome de Currarino, qui associe une malformation de l’intestin caudal dans 100% des cas (typiquement une sténose anorectale), une agénésie partielle du sacrum dans 90% des cas, et une tumeur présacrée dans 90% des cas (méningocèle, tératome, kyste neurentérique). Ce spectre malformatif découle d’une anomalie de la neurulation secondaire avec une mutation du gène HLXB9 retrouvée dans 50% des cas. Elle est à distinguer du TSC fœtal/néonatal isolé.
Le TSC néonatal bénin est également à distinguer des tumeurs germinales malignes de localisation sacrococcygienne qui sont diagnostiquées à un âge plus tardif, le plus souvent après 6 mois. Ces tumeurs malignes ont un développement endopelvien et représentent 40% des TSC diagnostiqués après l’âge de 1 an.
Seules les formes fœtales/néonatales isolées et donc bénignes seront traitées ici.
L’évolution naturelle des TSC impliquent trois enjeux distincts en fonction des périodes de la vie :
- En période périnatale, le pronostic vital du fœtus ou de l’enfant peut être mis en jeu en raison de phénomènes vasculaires.
- Dans la petite enfance, le pronostic devient oncologique, avec un risque de récidive (bénigne ou maligne), et un risque de dégénérescence spontanée du TSC qui justifie une exérèse chirurgicale systématique au diagnostic.
- A plus long terme, l’enfant peut être porteur de séquelles fonctionnelles, notamment digestives (constipation, troubles de la continence), urinaires (vessie neurologique, fuites urinaires), orthopédiques et cosmétiques.
 Diagnostic prénatal
Diagnostic prénatal
Dans 65 à 75% des cas, le TSC est diagnostiqué au cours des échographies de dépistage prénatales. Une masse du pôle caudal du fœtus est alors visualisée. Au diagnostic, la taille, la vascularisation doppler, la composition (solide, kystique, mixte), les biométries fœtales et la présence d’éventuelles complications sont appréciées. En l’absence de malformations associées ou d’antécédents familiaux, un examen cytogénétique n’est habituellement pas proposé.
Figure 2 : Coupe sagittale fœtale en échographie sur laquelle est visualisée une masse mixte du pôle caudal à prédominance exopelvienne (Altman I).
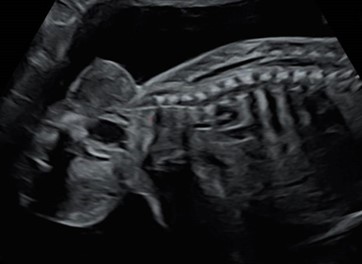
Photo Pr Yves VILLE
Une surveillance régulière est nécessaire au cours de la grossesse, notamment pour dépister la survenue de complications maternelles et/ou fœtales. La réalisation d’une IRM fœtale permet de confirmer le type de TSC selon la classification d’Altman, de dépister d’éventuelles complications et de rechercher une extension intra-spinale du TSC qui modifiera la prise en charge chirurgicale à la naissance et le pronostic [2].
Figure 3 : IRM fœtale (séquence T2 FIESTA) montrant un TSC mixte (*) responsable d’une mégavessie (V) et d’une hydronéphrose bilatérale (flèches).
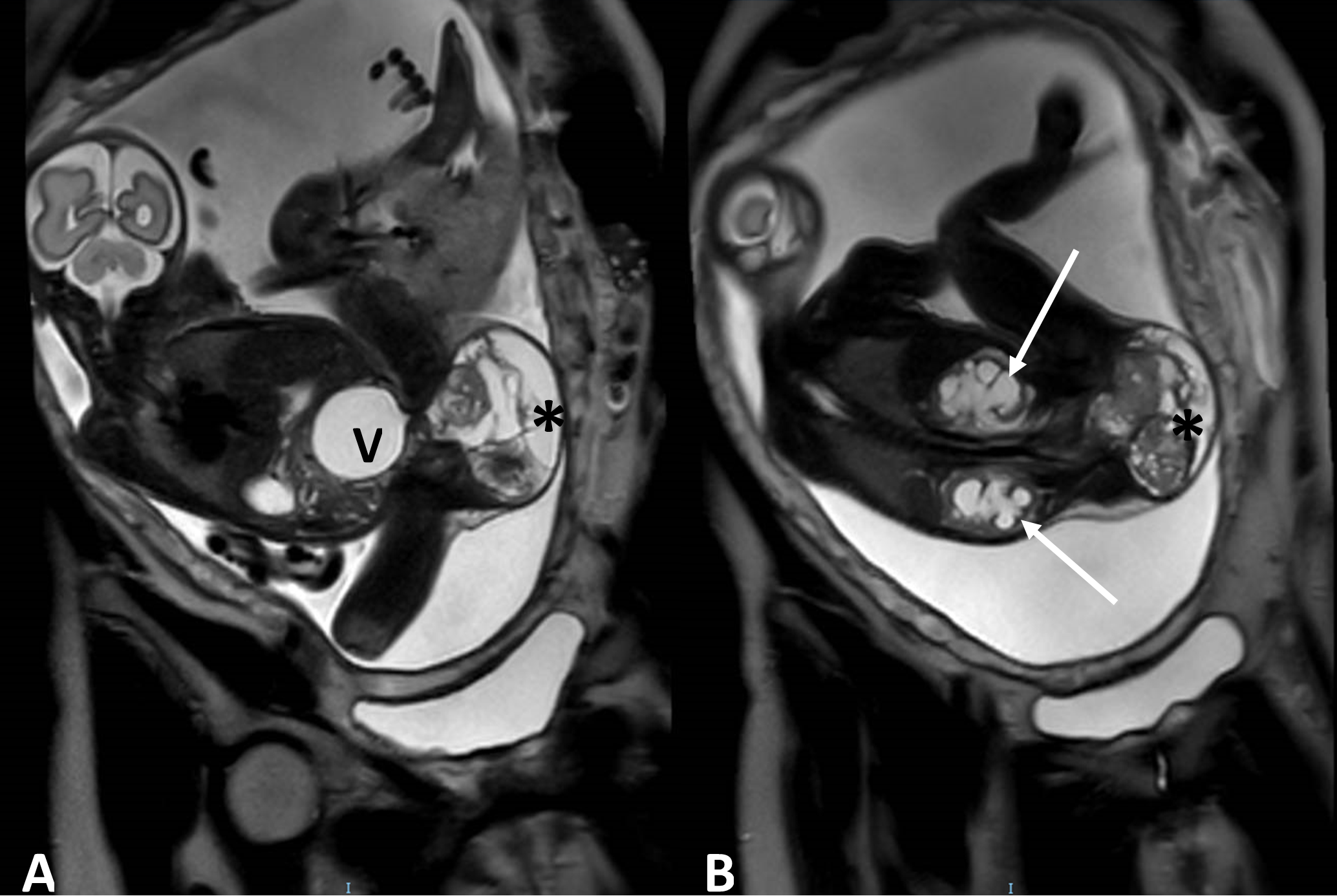
Photo Dr David GREVENT
Le tableau suivant résume les principales complications périnatales fœtales/néonatales et maternelles :
|
Complications fœtales/néonatales |
Complications maternelles et obstétricales |
|
. Insuffisance cardiaque à haut débit, liée à un shunt vasculaire intra-tumoral, pouvant entraîner une anasarque puis une mort fœtale in utero . Syndrome de masse, notamment sur le tractus urinaire, pouvant mener à une hydronéphrose bilatérale et/ou un tableau d’obstacle sous-vésical . Risque hémorragique, lié le plus souvent à une rupture tumorale (notamment perpartum) ou plus rarement à une hémorragie intra-tumorale spontanée . Prématurité |
. Syndrome en miroir en cas d’anasarque fœtale . Menace d’accouchement prématuré (distension utérine en cas d’hydramnios notamment) . Dystocie en cas d’accouchement voie basse . Complications liées à une cicatrice utérine, souvent corporéale en cas de tumeur volumineuse |
De nombreuses études se sont intéressées aux facteurs prénataux prédictifs d’une issue périnatale défavorable (mort fœtale in utero, décès néonatal, mise en jeu du pronostic vital néonatal). Celle-ci touche 20 à 30% des fœtus ayant un TSC de diagnostic anténatal. Les facteurs décrits comme associés à une issue défavorable sont : un terme précoce au diagnostic, une vitesse de croissance tumorale rapide (> 7mm/semaine), un volume tumoral (rapport entre volume tumoral et estimation du poids fœtal >0,12 avant 24 SA ou >0,16 quel que soit le terme), une tumeur à prédominance solide richement vascularisée [3,4].
En cas de complication hémodynamique, des signes d’insuffisance cardiaque à haut débit peuvent apparaître à l’échographie, tels qu’une cardiomégalie, un hydramnios, des anomalies des dopplers fœtaux (veine ombilicale, artère ombilicale, ductus venosus) et/ou une placentomégalie. Ces signes peuvent être accompagnée d’une anasarque fœto-placentaire, dont l’issue est létale en l’absence d’intervention.
Devant la présence de signes de complications hémodynamiques fœtales, la conduite à tenir dépend du terme d’apparition, de leur gravité et de leur évolutivité. Les différentes stratégies peuvent être une poursuite de la surveillance rapprochée, une extraction fœtale avec chirurgie d’exérèse à la naissance, ou une interruption médicale de grossesse si elle demandée par le couple. Des traitements in utero ont été décrits, soit pour diminuer la masse tumorale et donc l’effet shunt qui en résulte, soit comme traitement symptomatique d’une complication (amniodrainage, pose d’un drain vésico-amniotique en cas de compression du tractus urinaire, transfusion in utero), ou encore pour prévenir le risque dystocique (drainage antepartum d’une composante kystique importante).
Un accouchement en milieu chirurgical est nécessaire, même en l’absence de complication prénatale, pour permettre une évaluation rapide du TSC à la naissance et déterminer le délai optimal de la chirurgie d’exérèse. Un accouchement par césarienne est recommandé en cas de diamètre tumoral >5-7 cm selon les équipes.
La mortalité néonatale en cas de TSC de diagnostic anténatal est de l’ordre de 15%, avec notamment un risque hémorragique prédominant. Un volume tumoral important (>1L), la prématurité et la nécessité d’une résection en urgence ont été rapportés comme facteurs associés à ce risque de mortalité [5].
L’exérèse chirurgicale du TSC est systématique. Son délai dépend de la taille de la tumeur et de la tolérance de l’enfant, mais elle est habituellement réalisée à la naissance ou dans les premières semaines. L’exérèse doit être complète, idéalement en bloc et emportant le coccyx pour limiter le risque de récidive. Les deux principales complications postopératoires sont le défaut de cicatrisation (50% environ) et la rétention aigüe d’urines (10% environ).
 Risque oncologique dans la petite enfance
Risque oncologique dans la petite enfance
En raison du risque de récidive du TSC, une surveillance postopératoire est recommandée (tous les 3 à 6 mois pendant au moins 3 ans), et doit comprendre au minium un examen clinique et un dosage de l’aFœtoprotéine. Une échographie pelvienne peut également être réalisée. L’aFœtoprotéine permet de dépister les récidives maligne (tumeur du sac vitellin). Le taux de récidive décrit est compris entre 5 et 10%, avec 50% de récidives malignes et 50% de récidives bénignes. Les récidives surviennent habituellement dans les deux premières années. Un risque plus important de récidive a été rapporté chez les enfants porteur d’un TSC dont l’histologie est à prédominance immature [6].
Risque de séquelles fonctionnelles à long terme
Dans la littérature, en moyenne un tiers des patients opérés d’un TSC présentent des séquelles fonctionnelles postopératoires. Celles-ci peuvent être d’ordre digestif (constipation, souillures, absence de transit volontaire), urinaire (vessie neurologique, symptômes du bas appareil urinaire), cosmétique ou orthopédique.
Des séquelles digestives sont rapportées chez 20 à 30% des patients opérés d’un TSC. La constipation semble être le symptôme le plus fréquent (25-50%). Les facteurs décrits comme associés au risque de séquelles digestives sont principalement le volume tumoral (>9-10 cm), la présence de complications prénatales obstructives et la récidive tumorale.
Le taux de vessies neurologiques postopératoire varie beaucoup dans les études, entre 6 et 45% des enfants. Ce type de séquelles est définitif et peut être responsable d’infections urinaires à répétition et d’altération secondaire de la fonction rénale. Il est donc nécessaire de dépister les anomalies de vidange vésicale dans la petite enfance (échographie réno-vésicale avec mesure du résidu post-mictionnel) et de poursuivre les explorations en cas de symptômes d’anomalie de vidange, d’infection urinaire fébrile ou d’anomalie échographique. La présence de complications prénatales obstructives sur le tractus urinaire a été décrite comme associée à un risque de vessie neurologique, tout comme l’extension intra-spinale du TSC.
Les séquelles cosmétiques sont fréquentes (30% en moyenne) chez les patients opérés d’un TSC, et peuvent justifier de reprises chirurgicales. Le volume tumoral important semble être le facteur de risque identifié comme associé à ce type de séquelles. Une collaboration avec un chirurgien plasticien lors de la chirurgie d’exérèse peut être intéressante pour limiter ces séquelles.
Enfin, les séquelles orthopédiques sont plus rares (5-10% environ) et semblent plus fréquentes en cas d’extension intra-spinale de la tumeur.
Même si la présence de séquelles fonctionnelles est associée à une baisse de la qualité de vie, il semble que les adultes traités pour un TSC dans l’enfance ne présentent pas de différence significative en termes de qualité de vie par rapport à une population contrôle [7].
Références bibliographiques
- Peter Altman R, Randolph JG, Lilly JR. Sacrococcygeal teratoma: American Academy of Pediatrics Surgical Section survey-1973. J Pediatr Surg 1974;9:389–98. doi:10.1016/S0022-3468(74)80297-6.
- Avni FE, Guibaud L, Robert Y, Segers V, Ziereisen F, Delaet MH, et al. MR imaging of fetal sacrococcygeal teratoma: Diagnosis and assessment. Am J Roentgenol 2002;178:179–83. doi:10.2214/ajr.178.1.1780179.
- Vinit N, Benachi A, Rosenblatt J, Jouannic J-M, Rousseau V, Bonnard A, et al. Growth velocity of fetal sacrococcygeal teratoma as predictor of perinatal morbidity and mortality: multicenter study. Ultrasound Obstet Gynecol 2024;64:651–60. doi:10.1002/uog.29110.
- Gebb J, Khalek N, Qamar H, Johnson M, Oliver E, Coleman B, et al. High Tumor Volume to Fetal Weight Ratio Is Associated with Worse Fetal Outcomes and Increased Maternal Risk in Fetuses with Sacrococcygeal Teratoma. Fetal Diagn Ther 2018. doi:10.1159/000486782.
- Kremer MEB, Wellens LM, Derikx JPM, van Baren R, Heij HA, Wijnen MHWA, et al. Hemorrhage is the most common cause of neonatal mortality in patients with sacrococcygeal teratoma. J Pediatr Surg 2016;51:1826–9. doi:10.1016/j.jpedsurg.2016.07.005.
- van Heurn LJ, Derikx JPM, Hall N, Aldrink JH, Bailez MM, Chirdan LB, et al. Malignant transformation and tumour recurrence in sacrococcygeal teratoma: a global, retrospective cohort study. Int J Surg 2024;110:7177–86. doi:10.1097/JS9.0000000000002045.
- Kremer MEB, Dirix M, Koeneman MM, Van Baren R, Heij HA, Wijnen MHWA, et al. Quality of life in adulthood after resection of a sacrococcygeal teratoma in childhood:A Dutch multicentre study. Arch. Dis. Child. Fetal Neonatal Ed., vol. 100, 2015, p. F229–32. doi:10.1136/archdischild-2014-307589.